21 CFR Part 11 Compliance Guide
Nearly every FDA data integrity citation traces back to the same place: electronic records and signatures that weren’t managed in accordance with 21 CFR Part 11.
This guide covers what 21 CFR Part 11 actually requires, where regulated companies most often fall short, and what to look for in QMS software.
What Is 21 CFR Part 11?
21 CFR Part 11 is an FDA rule in Title 21 of the Code of Federal Regulations that makes electronic records and signatures as valid as paper ones. It was published in 1997 as FDA-regulated industries began transitioning from paper-based documentation to electronic systems.
| Subpart | Focus | Key Sections |
|---|---|---|
| Subpart A | General Provisions | Scope (11.1), Definitions (11.3) |
| Subpart B | Electronic Records | Closed system controls (11.10), Open system controls (11.30), Signature manifestation (11.50) |
| Subpart C | Electronic Signatures | General requirements (11.100), Signature components (11.200), ID codes and passwords (11.300) |
Part 11 does not replace the underlying FDA regulations that require records in the first place. It works alongside them. The underlying regulation (21 CFR Part 211 for pharmaceutical manufacturing, 21 CFR Part 820 for medical devices) tells you what records to keep and for how long. Part 11 outlines how to manage those records electronically so they remain trustworthy, tamper-proof, and legally equivalent to paper.
21 CFR Part 11 Requirements
1 Audit Trails
Under 11.10(e), every electronic record must have a secure, computer-generated, time-stamped audit trail that independently records the date and time of operator entries and any changes to records. Previously recorded data must remain visible and unaltered.
- Every record must capture who created or modified it, what changed, when, and why
- Audit trail entries cannot be altered or hidden by any user, including administrators
- Audit trail records must be retained for the same period as the underlying records
Manual system: Teams relying on manual audit trail documentation often maintain separate change logs or version history spreadsheets. These fail when investigators request the complete history of a specific record. Documentation is generally incomplete, inconsistently maintained, or stored separately from the record itself.
QMS software: Octave Reliance builds audit trails into every record across the platform.
Any update to a CAPA record, document, or deviation log is timestamped and permanently associated with that record. Investigators can pull a complete, tamper-proof change history for any record in the system.
2 Access Controls
Under 11.10(d) and 11.300, access to systems that contain Part 11 records must be limited to authorized users through individual IDs and passwords.
- Every user must have a unique, non-shared login
- When personnel leave, access must be revoked and documented
- The system must detect and respond to unauthorized login attempts
- They system must control password issuance, including procedures for lost or stolen credentials
Manual system: Access control policies enforced through IT infrastructure alone often fall short. When personnel turnover happens quickly, it can delay account deactivation. Additionally, shared accounts are common in production environments where logging in and out between shifts feels slow and inefficient.
QMS software: Purpose-built QMS platforms enforce role-based permissions. Account deactivation workflows tie to HR processes, so terminated employee access is removed systematically rather than relying on manual IT tickets.
3 Electronic Signatures
Under 11.100 and 11.200, electronic signatures must meet specific requirements:
- Each signature must be unique to one individual and cannot be reused or reassigned
- Non-biometric signatures require two identification components (user ID + password) on first signing; at least one on subsequent signings in the same session
- Every signed record must display the signer’s name, timestamp, and signature meaning (approved, reviewed, authorized, etc.)
- Companies must certify intent to the FDA in writing before or at first use, confirming signatures are legally binding and equivalent to handwritten signatures
Manual system: Wet ink signatures on printed records technically avoid Part 11 requirements. That said, they reintroduce the operational costs that Part 11-compliant electronic systems eliminate: manual routing for signatures, physical storage, slower approval cycles, and the risk of lost or incomplete signature pages.
QMS software: MasterControl requires two separate password components at the signature level: one for login and a second for document approval.
It appends the signer’s name, timestamp, and signature meaning to every signed record. The system’s audit trail fully captures signature chains for batch releases and CAPA closures.
4 System Validation
Under 11.10(a), any system that creates, modifies, maintains, or transmits Part 11 records must be validated to ensure it works consistently and accurately. It should also be able to detect invalid or altered data.
The FDA finalized updated Computer Software Assurance (CSA) guidance in September 2025. This replaced the prescriptive Computer Software Validation (CSV) framework with a risk-based approach. Validation effort should now scale with the impact the system has on product quality and record integrity.
Key validation requirements include:
- Documented evidence that the system performs as intended, prior to regulated use
- Re-validation when software is updated, reconfigured, or business use changes materially
- Validation documentation retained for the life of the system
Manual system: Many companies completed a validation at initial system deployment and have not revisited it since. Software updates, configuration changes, and new use cases since the original validation create gaps that are difficult to defend under inspection.
QMS software: QT9 QMS ships pre-validated with vendor-executed IQ/OQ/PQ documentation included out of the box. QT9 updates this documentation with every software release at no extra charge.
This matters under the FDA’s 2025 CSA guidance, which treats validation as an ongoing obligation rather than a one-time project.
5 Record Integrity and Retention
Under 11.10(b) and 11.10©, electronic records must be protected against unauthorized alteration. Accurate copies must be retrievable throughout the entire retention period required by the applicable predicate rule:
- Records must be reproducible in both human-readable and electronic format
- Records must be retrievable throughout the full retention period; for some pharmaceutical records, shelf life plus additional years
- Backup and recovery procedures must be tested and documented
Manual system: Paper-based backup systems create audit exposure when investigators request records from 5 or 10 years ago, and documentation is incomplete or difficult to retrieve. Digital backups without tested recovery procedures run a similar risk.
QMS software: QAD EQMS integrates quality processes with QAD’s ERP, connecting document control, CAPA, and audit workflows across the enterprise.
Electronic document routings and approvals ensure records remain current and retrievable, and the system supports ISO 13485 and 21 CFR Part 820 compliance for regulated manufacturers.
6 Operational Controls and Personnel Accountability
Under 11.10(i) and 11.10(k), Part 11 requires that personnel who develop, maintain, or use electronic systems have the education, training, and experience to perform their assigned tasks. Companies must also establish written policies that hold individuals accountable for actions related to electronic records and signatures.
- Training records must show employees were trained on electronic documentation protocols before system use
- SOPs must govern system use, signature application, access management, and record retention
- If something goes wrong with a record, documentation must enable identification of who was responsible and what happened
Manual system: Training matrices and SOP binders can satisfy this requirement, but only if kept current. An SOP that governs a version of the software that no longer exists, or a training record that doesn’t reflect the current workflow, creates an audit gap.
QMS software: Integrated training management modules–such as those in Octave Reliance–automatically flag retraining requirements when SOPs are updated. Every employee’s training status against the current document versions is tracked and reportable on demand.
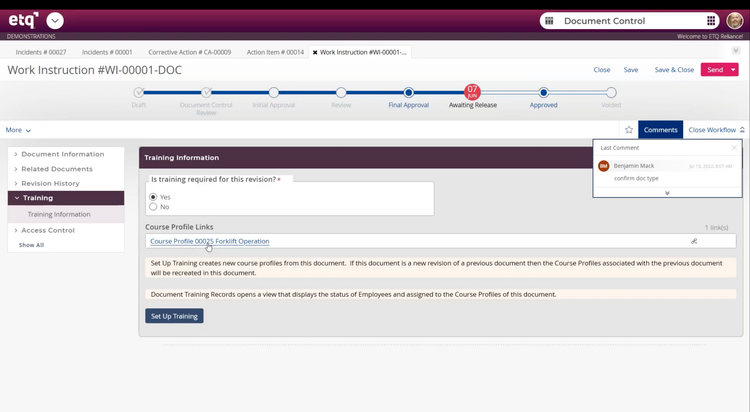
Predicate Rules and Part 11
Which systems require Part 11 compliance depends on predicate rules: the FDA regulations that require specific records to exist in the first place. Part 11 applies only to records that a predicate rule already requires you to maintain, like batch records, deviation reports, and CAPA documentation. If a record isn’t required by any predicate rule, Part 11 doesn’t apply, even if it’s stored digitally.
The FDA clarified this in a 2003 guidance document that narrowed Part 11’s scope. Practical test: if a regulated decision is being made from an electronic record, Part 11 applies. If the team prints it and relies on the paper version, it may not.
Open Systems vs. Closed Systems
Part 11 distinguishes between closed systems, where access is controlled by the record owners, and open systems, where that control is not maintained by the record owners. This might be a cloud platform where suppliers, CDMOs, or CROs log in directly.
Open systems require all closed-system controls, plus additional measures such as data encryption at rest and in transit, and digital signature methods. As more regulated companies move to cloud platforms with external party access, this distinction has become practically important.
Who Does 21 CFR Part 11 Apply To?
Part 11 applies to any company regulated by the FDA that uses electronic systems in place of paper records to meet regulatory requirements. That includes:
- Pharmaceutical manufacturers (regulated under 21 CFR Parts 210 and 211)
- Medical device manufacturers (regulated under 21 CFR Part 820)
- Biologics and biosimilar manufacturers
- Contract manufacturers (CDMOs) and contract testing laboratories
Enforcement Trends in Pharma
FDA enforcement of Part 11–primarily through data integrity citations–has intensified significantly. The FDA issued 190 warning letters to drug and biologics firms in FY2024, with CDER warning letters jumping 50% in FY2025.
The consequences extend well beyond a warning letter: product recalls cost $10 million to $100 million, depending on scope. FDA consent decrees have cost companies hundreds of millions in remediation and lost production capacity.
For pharmaceutical companies, violations most frequently cited involve quality unit failures under 21 CFR 211.22, CAPA inadequacies, and data integrity issues, all directly tied to Part 11 compliance.
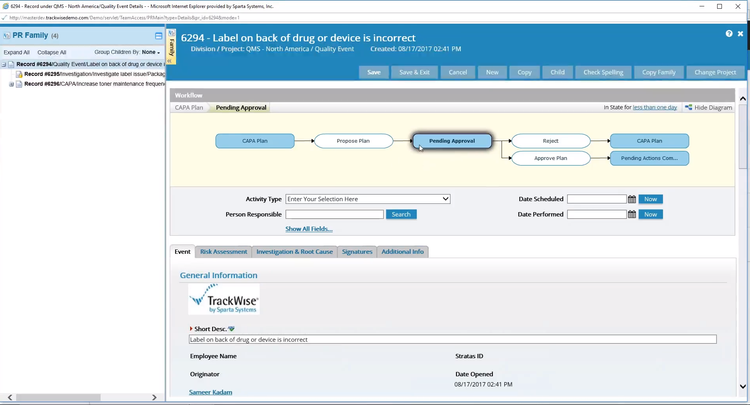
Enforcement Trends in Medical Devices
For medical device manufacturers, Part 11 has taken on new urgency with the FDA’s Quality Management System Regulation (QMSR), effective February 2026. QMSR aligns 21 CFR Part 820 with ISO 13485.
Device companies managing both frameworks now face heightened scrutiny on electronic records and signatures across design controls, CAPA, and production documentation.
Medical device QMS software, designed with Part 11 and ISO 13485 in mind, significantly reduces the audit preparation burden compared to systems that treat these standards as separate exercises.
Where Companies Get It Wrong
Most Part 11 audit findings fall into a small cluster of recurring failures.
Shared accounts. One login shared across a shift because individual logins feel impractical. Under Part 11, an electronic signature must trace back to one identifiable individual. A shared login makes that impossible. If three employees used the same account when a batch was approved, you cannot prove who authorized it.
Shadow systems. The validated system is Part 11 compliant, but someone is tracking open CAPAs in a spreadsheet because the official system is slower. The moment regulated decisions are made from that spreadsheet, it becomes a Part 11 record and is not compliant.
Stale validation documentation. A system was validated at deployment. Since then, software has been updated, workflows reconfigured, and new modules added. The original documentation no longer reflects how the system actually operates. Under the FDA’s 2025 CSA guidance, validation is an ongoing activity, not a one-time project.
Misidentifying which systems are subject to Part 11. Companies over-validate systems that don’t hold predicate rule records or fail to validate systems that do. The question for every electronic system: does it create, modify, maintain, or transmit records that an FDA predicate rule requires you to keep? If yes, Part 11 applies.
21 CFR Part 11 Compliance Software
The right system enforces controls automaticall, including audit trails, access management, electronic signatures, and validation documentation. When evaluating QMS software for Part 11, the questions that matter most are:
- Is Part 11 compliance built into the platform’s core design, or is it a configurable add-on?
- Does the vendor provide IQ/OQ/PQ validation packages? Are those packages updated with software releases?
- Can the system provide complete, human-readable copies of all records on demand for an FDA investigator?
- How does the system handle external party access (open system controls)?
These are our top picks for Part 11-compliant QMS software across pharmaceutical and medical device applications: