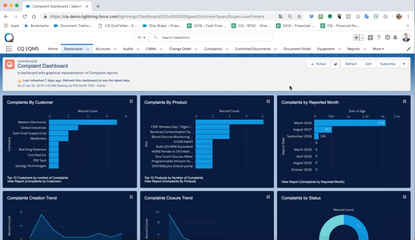
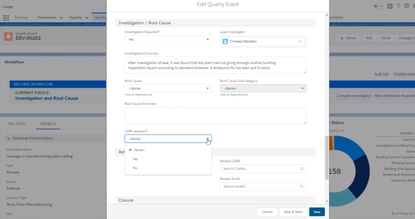
CAPA in the Pharmaceutical Industry
CAPA deficiencies appear in roughly 40% of Form 483 observations issued by the FDA to pharma companies. This guide breaks down what CAPA in the pharmaceutical industry actually requires. It also shows where businesses most often fail and how QMS software can help you stand up under regulatory scrutiny.
What Is CAPA in the Pharmaceutical Industry?
CAPA (Corrective and Preventive Action) in the pharmaceutical industry is a structured process for investigating quality issues, identifying root causes, and preventing recurrence. It sits at the center of the pharmaceutical quality system and is often treated by regulators as a proxy for overall quality system health.
At a basic level:
- Corrective action addresses the root cause of an identified issue.
- Preventive action addresses potential issues before they occur.
Need to find a QMS system? Check out our best pharmaceutical QMS page to explore our full list of options.
Regulatory Framework: What the FDA and Global Bodies Expect
Pharmaceutical CAPA requirements are distributed across multiple frameworks:
| Regulation / Guideline | What It Covers for CAPA |
|---|---|
| 21 CFR Part 820.100 | Device-focused CAPA regulation; widely used as the “gold standard” model |
| 21 CFR Part 211.192 & 211.180(e) | Failure investigations and annual product review requirements |
| ICH Q10 | Establishes CAPA as a core element of the pharmaceutical quality system |
| ICH Q9 | Introduces risk-based prioritization for CAPA activities |
| EU GMP (EudraLex Volume 4) | Chapters 1 and 8 emphasize CAPA, investigations, and quality systems |
| ISO 9001:2015 & [ISO 13485]( | Define formal CAPA structures; relevant for combination products |
| PIC/S Guidance | Harmonized global GMP expectations, including CAPA practices |
| 21 CFR Part 11 | Requirements for electronic records, audit trails, and electronic signatures |
| QMSR | Aligns Part 820 more closely with ISO 13485 standards |
The key takeaway: the FDA does not prescribe a specific CAPA template for pharma. Companies must define their own processes, but regulators will evaluate whether they are risk-based, documented, and consistently executed.
What Triggers a CAPA?
A CAPA should be opened due to:
- Deviations and nonconformances
- Out-of-specification (OOS) or out-of-trend (OOT) results
- Customer complaints or adverse events
- Internal or external audit findings
- Supplier quality issues
- Regulatory inspection observations (including Form 483)
- Recurring issues identified through trend analysis Why CAPA matters
- Patient safety: Prevents contamination, dosing errors, and ineffective products
- **Compliance: Reduces risk of Form 483s and warning letters
- Cost & improvement: Cuts rework and drives product quality gains Not every issue requires a full CAPA. Mature systems apply risk-based criteria to determine whether an event warrants a formal CAPA versus a correction or investigation alone. This decision logic is frequently scrutinized during inspections.
The CAPA Process: Step by Step
A typical CAPA process in pharma follows a structured lifecycle:
- Issue identification: Capture deviations, nonconformances, complaints, or audit findings.
- Risk assessment and prioritization: Evaluate severity, frequency, and detectability to determine if a CAPA is required.
- Correction and containment: Implement immediate actions to control the issue (e.g., batch quarantine). This is not the same as corrective action.
- Root cause analysis (RCA): Use structured methods (5 Whys, Fishbone, FMEA) to identify the true cause—not just symptoms.
- CAPA planning and implementation: Define and execute both corrective and preventive actions, including process updates, training, or system changes.
- Verification: Verification of implementation (VOI) - Confirm actions were executed as planned; verification of effectiveness (VOE): Demonstrate the actions actually resolved the issue.
- Closure and documentation: Compile complete documentation with traceability from issue to resolution.
- Management review and trend analysis: Aggregate CAPA data to identify systemic issues and inform continuous improvement.
A common inspection failure is confusing VOI with VOE: closing CAPAs after confirming execution without proving effectiveness.
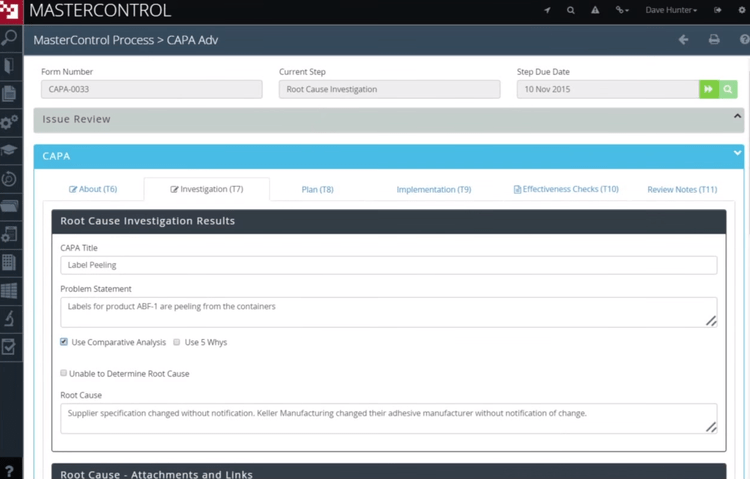
Common CAPA Pitfalls in Pharma
Most CAPA failures aren’t due to missing procedures—they stem from execution gaps.
One of the most frequent issues is shallow root cause analysis. Teams default to “human error” without investigating systemic contributors like process design, training gaps, or equipment issues. Regulators consistently flag this as a superficial approach.
Another common breakdown is confusing correction with corrective action. Fixing a single batch issue without addressing why it occurred leads to repeat deviations and findings.
Effectiveness verification failures are also quite common. CAPAs are closed without objective evidence that the solution worked, which is one of the most cited issues in Form 483 observations.
Many organizations also struggle with inconsistent CAPA initiation criteria. Without clear thresholds, some issues escalate unnecessarily, while others that warrant CAPA are missed entirely.
Operationally, CAPA systems often become siloed. When QA owns CAPA without input from manufacturing, engineering, or regulatory, this results in incomplete root causes and narrow solutions.
Finally, weak systems lack traceability and trending. Poor documentation, missing audit trails, and failure to analyze patterns across CAPAs prevent organizations from identifying systemic risks.
What to Do When FDA Cites Your CAPA System
When a Form 483 or warning letter cites CAPA deficiencies, response quality matters as much as the fix itself. Regulators expect structured, risk-based responses.
- Respond within 15 business days
- Clearly acknowledge the issue and scope
- Avoid superficial fixes and focus on systemic remediation
- Provide timelines, milestones, and measurable outcomes
- Commit to progress updates where appropriate
- Ensure all actions are fully documented with traceability
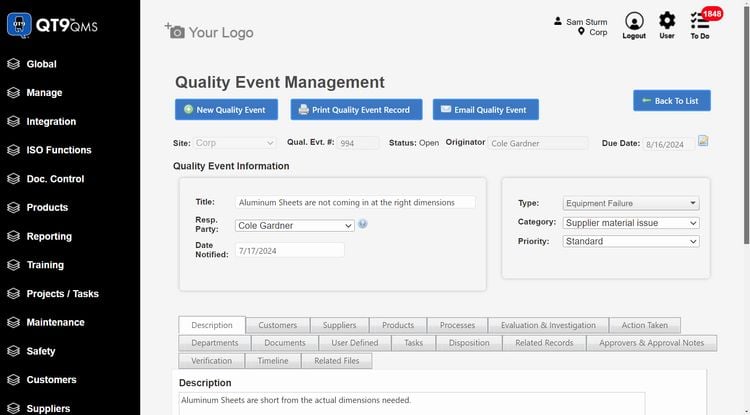
Building a Mature CAPA System
Mature CAPA systems evolve from reactive issue handling to proactive quality management.
Best practices
- Integrate CAPA with deviations, complaints, change control, and training
- Apply risk-based prioritization using ICH Q9 principles
- Track metrics like closure time, recurrence rate, and aging
- Establish cross-functional CAPA review boards
- Perform internal CAPA audits regularly
- Use data to identify patterns before they escalate
You can evaluate CAPA software or modern eQMS platforms to help automate these processes.
How eQMS software supports CAPA
QMS software enables more consistent and scalable CAPA execution, including automated workflows and approvals and centralized documentation with version control. Common platforms include MasterControl, TrackWise, Qualio, Veeva Vault QMS, ComplianceQuest, and QT9.