Design Controls for Medical Devices Guide
Design controls for medical devices are systematic procedures that ensure manufactured devices meet users’ needs, intended use, safety, and regulatory standards throughout development. They’re mandated by FDA’s QMSR (formerly 21 CFR 820) and international standards like ISO 13485, which are now more closely aligned than ever.
This guide walks through the full design controls process, from regulatory framework to each of the nine required elements, common audit findings, and the QMS software that helps teams stay compliant.
Who Has to Comply?
Design controls are mandatory for all Class II and Class III medical devices under FDA jurisdiction. Most Class I devices are exempt, but there are six specific exceptions where design controls still apply: devices automated with computer software, tracheobronchial suction catheters, surgeon’s gloves, protective restraints, manual radionuclide applicator systems, and radionuclide teletherapy sources.
Outside the U.S., the picture is similar in substance if not in structure. ISO 13485:2016 requires design and development controls for any organization involved in designing medical devices, regardless of geography. The EU Medical Device Regulation EU MDR 2017/745 requires design controls as part of its conformity assessment process. If you sell devices in more than one market, you’re effectively subject to all three frameworks, and the recent QMSR harmonization makes that easier to manage from a single quality system.
The Regulatory Landscape
As of February 2, 2026, the FDA and international standards are more closely aligned than ever. Here’s how the pieces fit together.
QMSR: The Current U.S. Framework
The FDA’s QMSR took effect on February 2, 2026, replacing the old QSR. This is a huge change to how manufacturers are regulated, and everyone who manufactures finished devices marketed in the US must now comply.
The QMSR amends 21 CFR Part 820 by incorporating ISO 13485 by reference. Part 820 still exists and is still the legal authority. If an auditor cites you, they’re citing Part 820. But most of the old prescriptive language, including the design control requirements previously spelled out in 820.30, has been replaced with references to the corresponding sections of ISO 13485:2016. For design controls, that means Clause 7.3.
There were many other changes, including control of records, risk-based thinking, and the retirement of the Quality System Inspection Technique (QSIT). If you want to read more about QMSR, learn more here: FDA QMSR Explained: What Changed and What It Means for Your QMS.
ISO 13485: 2016, Clause 7.3
The international standard for medical device quality management systems is now also the backbone of U.S. design control requirements under QMSR. Clause 7.3 covers design and development: planning, inputs, outputs, reviews, verification, validation, transfer, and change control. If you’re building a QMS from scratch, frame it around ISO 13485. It satisfies both international and U.S. requirements in a single framework.
EU MDR 2017/745
The European Medical Device Regulation requires a QMS covering all aspects of product realization, including design controls. Compliance is achieved by following ISO 13485’s design and development process. EU MDR also requires clinical evaluation and post-market surveillance to be integrated into the design lifecycle.
The Design Control Process
The most widely recognized visual for design controls is the FDA’s waterfall diagram, originally published in the 1997 Design Control Guidance for Medical Device Manufacturers. It shows user needs flowing into design inputs, which are translated into design outputs through a design process. Verification links outputs back to inputs, and validation links the finished device back to user needs. Design reviews gate each transition.
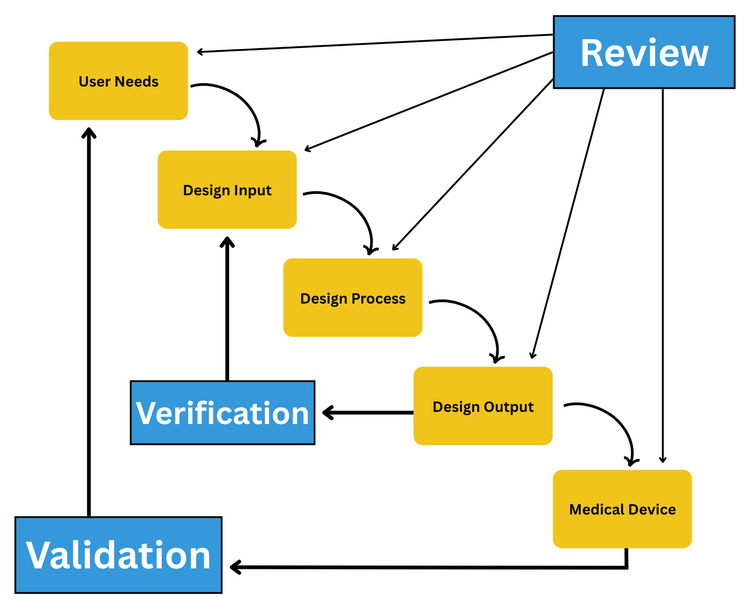
The 9 Elements of Design Controls
1 Design and Development Planning
Each manufacturer must establish and maintain plans that describe the design activities, cross-functional interfaces, and implementation responsibilities. A good plan documents what you’ll design, how you will execute and review each stage, who owns each activity, and the planned V&V activities.
Common mistake: Treating the design plan as a static document written once at kickoff. The regulation requires plans to be updated as the design evolves. A plan that doesn’t reflect the current state of the project won’t hold up in an audit.
2 Design Input
Design inputs translate user needs into specific, measurable, testable requirements that engineers can design against. They typically span functional, performance, safety, regulatory, human factors, and environmental categories. This phase often consumes roughly 30% of total project time, and for good reason. Incomplete inputs cascade through the entire development process.
Common mistake: Conflating user needs with design inputs. User needs are qualitative (“easy to hold”). Design inputs are engineering specs derived from those needs (“shall weigh no more than 200g with a grip diameter between 25-35mm”). Keeping them separate with documented traceability is best practice.
3 Design Output
Design outputs are the documented results of the design effort: specifications, drawings, CAD files, code, schematics, material specs, labeling, packaging, and instructions for use. Every output must trace back to one or more inputs and include acceptance criteria that define conformance. Don’t forget that packaging, labeling, and IFU are part of the device and subject to the same controls.
Common mistake: Outputs that omit acceptance criteria, making verification impossible.
4 Design Review
Conduct formal, documented evaluations at defined stages to assess whether the design meets requirements and to identify problems. You must include representatives from all relevant functions, plus at least one independent reviewer who is not directly responsible for the design. Most organizations perform reviews at major phase transitions, but frequency depends on device complexity and risk.
Common mistake: Companies that rubber-stamp reviews with no documented action items or follow-up.
5 Design Verification
“Did we design the device right?” Verification confirms that design outputs meet design input requirements through objective evidence, using methods like inspection, testing, analysis, and demonstration. Each activity should be protocol-driven with pre-defined acceptance criteria and documented results.
Common mistake: No pre-defined acceptance criteria, or criteria so vague they can’t meaningfully pass or fail.
6 Design Validation
“Did we design the right device?” Validation confirms the finished device meets user needs and intended uses under actual or simulated conditions. This is the most frequently cited design control element in FDA 483 observations. In FY2020, design validation accounted for 48 of the 178 design control citations issued by the FDA.
The regulation is explicit: validation must use production-equivalent units, not prototypes. It must also include software validation and risk analysis where appropriate, confirming that risk controls identified through ISO 14971 are effective.
Common mistake: Validating prototypes instead of production-equivalent devices.
7 Design Transfer
Design transfer ensures the design is correctly translated into production specifications. This is the most commonly glossed-over element, both in guides and in practice. A common failure mode is design “thrown over the wall” to manufacturing without complete documentation of process specs, supplier qualifications, or personnel training.
Effective transfer involves finalizing manufacturing process specifications, completing process validation, establishing purchasing controls, qualifying suppliers, and training production personnel. The best practice is involving manufacturing early in design reviews, not just at handoff.
Common mistake: Treating transfer as a single handoff event rather than an ongoing process.
8 Design Changes
All design changes after the initial design review must be identified, documented, assessed for impact, verified or validated as appropriate, reviewed, and approved before implementation. If a change affects a previously verified or validated aspect, those activities may need to be repeated. Changes that significantly impact safety, effectiveness, or intended use could trigger a new 510(k).
Common mistake: Businesses often have undocumented changes during development. Teams move fast and plan to “document later,” which never comes.
9 Design History File (DHF)
The DHF is the complete record demonstrating that the design was developed per the approved plan and regulatory requirements. It’s the auditor’s first stop. It should tell the full story of how the device went from concept to production, including the rationale behind key decisions. It’s important to distinguish the DHF from two related records:
| Record | Purpose | Contains |
|---|---|---|
| DHF (Design History File) | How the device was designed | Plans, inputs, outputs, reviews, V&V, changes |
| DMR (Device Master Record) | How to manufacture the device | Drawings, specs, procedures, acceptance criteria, labeling |
| DHR (Device History Record) | Proof a unit/lot was manufactured correctly | Production records, inspection results, batch records |
Design transfer is where DHF outputs become DMR-ready content. After transfer, DHRs are created for each production run.
Common mistake: Treating the DHF as a document dump with no traceability. A well-maintained DHF traces every requirement to evidence that it was met.
Common FDA 483 Findings for Design Controls
Understanding what auditors actually cite helps focus your compliance efforts where they matter most.
- Validation is the #1 citation: Design validation was cited 48 times in FY2020 alone, far exceeding any other design control clause. The most frequent issues are having no validation procedures at all, using prototypes instead of production-equivalent units, and omitting risk analysis from validation activities.
- Broken traceability and documentation: Auditors regularly find DHFs with no clear linkage between design inputs, outputs, and verification/validation results. They also find design changes that are made without formal review or approval. Design reviews that were either skipped entirely or conducted without the required independent reviewer.
- The cost is real: The cross-sectional study referenced earlier found device design accounted for 31.4% of Class I cardiovascular recalls. Most of these findings stem from fragmented documentation, broken traceability, and manual processes, which are exactly the problems that purpose-built QMS software is designed to solve.
How QMS Software Supports Design Controls
Many medical device teams start with spreadsheets, shared drives, or tools like Google Drive to manage design control documentation. This works when you’re just starting out, but it falls apart as you grow. The moment your team scales beyond a few people, the number of documents multiplies, or an auditor asks you to demonstrate traceability from a user need through verification, manual systems fall apart.
Design controls generate an enormous volume of interconnected documentation: plans, requirements, specifications, protocols, reports, risk files, review records, and change orders. So, beyond just organizing these documents, QMS enforces the connections between them. That way, traceability doesn’t depend on someone remembering to update a spreadsheet.
Leading QMS Platforms
Several platforms are built specifically for medical device design controls, or have strong modules dedicated to them:
Greenlight Guru
Greenlight Guru is purpose-built for medical device companies and is widely used by startups and growth-stage manufacturers. It provides built-in traceability between design inputs, outputs, verifications, and risk controls, with templates aligned to both QSR and QMSR terminology.
Qualio
Qualio is popular with startups and small teams for its fast implementation and intuitive interface. It offers core design control workflows, document management, training, and CAPA in a single platform with compliance support for ISO 13485 and FDA 21 CFR Part 820.
MasterControl
MasterControl is an enterprise-grade platform used by large pharmaceutical and medical device organizations. It offers electronic device history records (eDHR), strong document control, and deep integration with ERP and manufacturing systems.
QT9 QMS
QT9 QMS provides a validated, cloud-based platform with a dedicated design controls module. It supports FDA, ISO 13485, and EU MDR requirements and is positioned for companies that want comprehensive QMS coverage without enterprise-level complexity.
uniPoint QMS
uniPoint offers a modular QMS with design control capabilities alongside document control, CAPA, audit management, and supplier management. It’s used by manufacturers across multiple industries, including medical devices.