ISO 13485 Compliance: How Medical Device Teams Maintain It
ISO 13485 medical device compliance looks achievable on paper at first–but it all fails when you can’t explain gaps to an auditor.
We help manufacturers evaluate medical device QMS platforms every day, and the same failures surface again and again.
This guide focuses on where ISO 13485 compliance breaks down in the real world–and what you can do now to prevent that from happening, as your company evolves.
What Is ISO 13485?
ISO 13485 is an international quality management system (QMS) standard for the medical device industry. It applies across the full product lifecycle, including:
- Design & development
- Production
- Storage
- Distribution
- Installation
- Servicing
| Clause | Title | What Auditors Focus On |
|---|---|---|
| 1 | Scope | Applicability of the standard to your operations |
| 2 | Normative References | Use of referenced standards (e.g., ISO 14971) |
| 3 | Terms and Definitions | Consistent use of terminology |
| 4 | Quality Management System | Document control, system integrity, records |
| 5 | Management Responsibility | Accountability, management review, oversight |
| 6 | Resource Management | Training, infrastructure, competence |
| 7 | Product Realization | Design controls, traceability, production |
| 8 | Measurement, Analysis, & Improvement | CAPA, internal audits, continuous improvement |
FDA’s QMSR framework (effective 2026) incorporates ISO 13485 into 21 CFR Part 820 for medical device QMS. In other words, FDA inspections and ISO audits are becoming more aligned, so don’t think of it as a one-time certification exercise. Consider it an ongoing compliance practice.
ISO 9001 vs ISO 13485
Whereas ISO 9001 is a broad quality management standard applicable to any industry, ISO 13485 focuses specifically on medical device organizations.
ISO 9001 audits focus on verifying continual improvement, non-conformities, and QMS effectiveness. ISO 13485 audits want you to demonstrate whether your QMS conforms to medical device regulations, design controls, supplier oversight, and risk management.
Where ISO 13485 Audits Go Wrong
Auditors do not assess ISO 13485 clause by clause: they follow evidence trails. For example:
- A document leads to a training record
- A training record points to a design change
- A design change triggers a risk assessment
- A risk assessment leads to a corrective action
- A corrective action leads to management oversight
Most ISO 13485 audit findings center around a small cluster of operational breakdowns. We’ll cover:
- The most common types of failures
- How to prevent them with manual systems
- How to automate compliance with a QMS
ISO 13485 Document Control
Clause 4.2: This is one of the first areas auditors probe. To meet ISO 13485 documentation requirements, teams often depend on shared drives, file naming conventions, and folders–but this is where version sprawl begins to creep in over time.
Common audit issues include:
- Missing or opaque approvals
- Obsolete documents still accessible
- Different SOP versions in circulation
Manual system: This requires a master document list, revision tracking, a locked obsolete folder, and tight permissions. However, a single misfiled document can undermine the entire system.
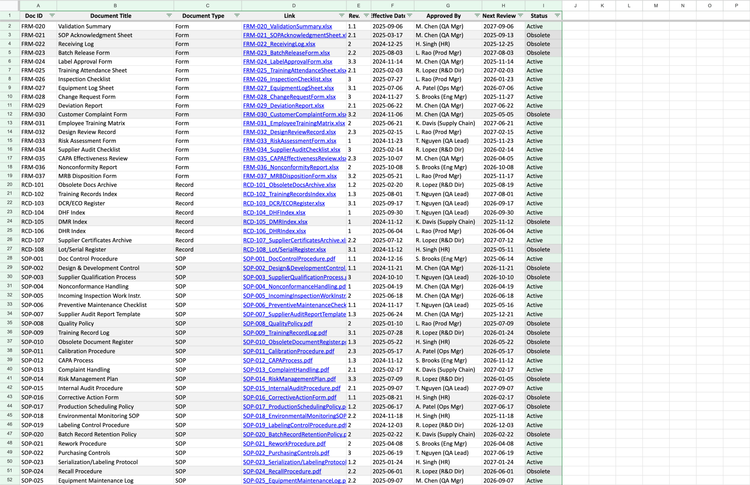
QMS software: Unifize EQMS allows you to view each procedure living in its own thread. If someone needs to review or approve a change, it’s available all in one view. Additionally, the system automatically logs updates and timestamps signatures. Older versions get marked “obsolete,” so no one on your team can accidentally use them.
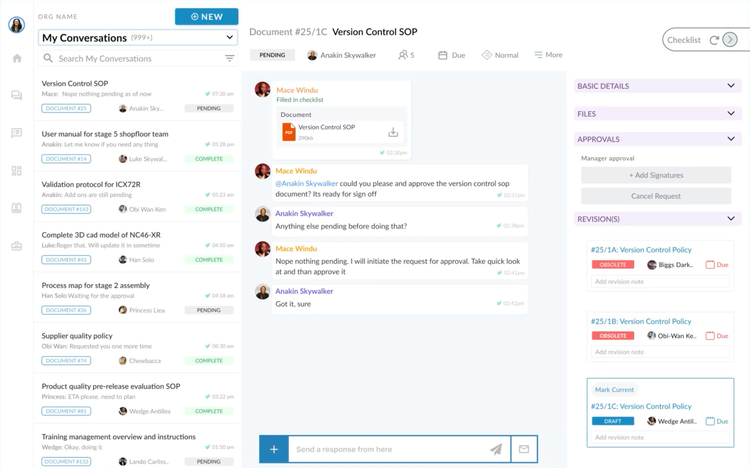
Training Requirements & Human Resources
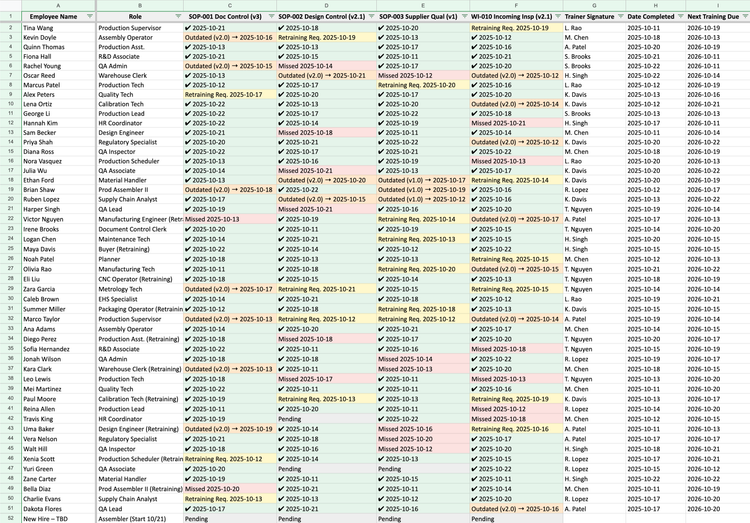
Clause 6.2: ISO 13485 requires more than just proof that training happened. Auditors need evidence that every employee was trained on the correct version of each procedure.
Manual system: You’ll need a training matrix with dated signatures for each session and a copy of the exact SOP revision they were trained on. Failures arise because updating a procedure means retraining everyone and tracking who’s current on which version–without missing any one individual.
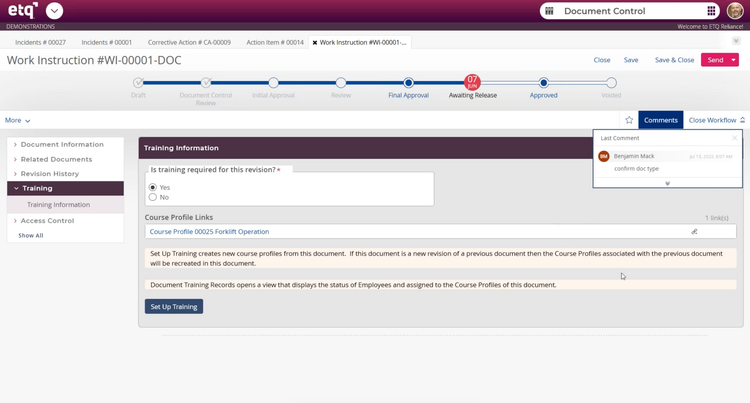
QMS software: EtQ Reliance triggers a training flag anytime work instructions are updated in the system. It displays the course profile and auto-assigns retraining to every employee who needs it.
Design Controls
Clause 7.3: A major part of ISO 13485 compliance is design control, especially when products evolve. Here, the auditor wants to see a clear line from design inputs to design outputs, verification, and validation. They also want to see that every change was reviewed, approved, and documented.
Manual system: This requires keeping folders for each design phase with signed and dated approvals. Where this gets difficult is when you’re manufacturing multiple medical devices. The auditor asks for your design history file (DHF), and you’re piecing together design data from five different places, like emails and slide decks.
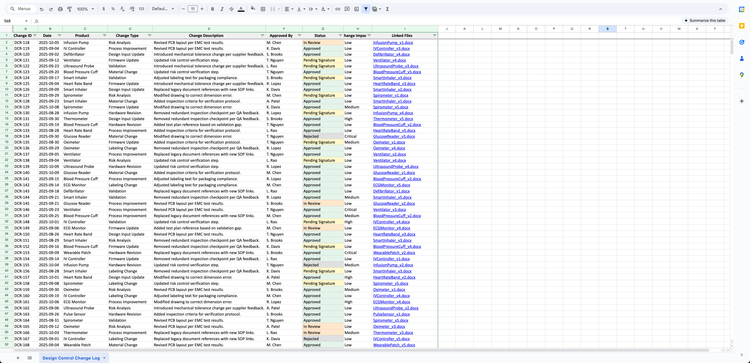
This is also where ISO 13485 risk management comes into play. Under ISO 14971, design changes require reassessment of risk. Risk files that exist but aren’t updated to reflect design changes often trigger follow-up findings.
QMS software: QT9 QMS lets you build DHF templates and tailor each plan to show what matters, including risk assessments, sign-offs, and verification. Because DHRs, DHFs, and DMRs are all connected, you’ve got a complete record from design through production.
ISO 13485 Corrective Action
Clause 8.5: Corrective and preventive action (CAPA) is where auditors revisit past issues. They find nonconformances from two years ago and want to see your root cause analysis–and verification that the fix actually resolved the problem. Many teams track ISO 13485 CAPA in spreadsheets, which don’t enforce rigor. Root cause quality can vary, and effectiveness checks may get skipped.
CAPA also intersects with:
- ISO 13485 risk management: when nonconformances surface unmitigated risks
- ISO 13485 supplier management: when issues originate externally
Manual system: You’ll need a form with mandatory fields for problem description, root cause, corrective action, owner, due date, and an effectiveness check with evidence. You’ll also need a separate spreadsheet that tracks the status of each CAPA form.
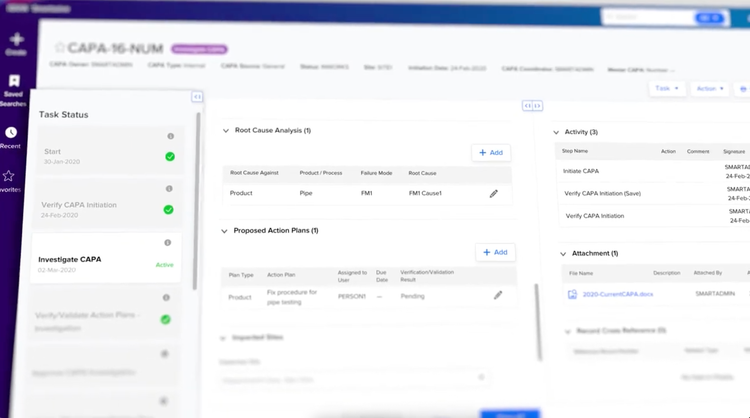
QMS software: SmartSolve shows you a structured CAPA workflow onscreen: root cause analysis, proposed action plans, all tracked in a task status view. It guides you through each step and maintains a complete audit trail all the way, so you never have to reconstruct anything from memory.
Traceability Requirements
Clause 7.5.9: Traceability is often the most stressful part of an audit. Customer complaints, recalls, or sampling exercises will lead auditors to ask:
- Where did the component originate?
- How was it used in production?
- Which customers received affected units?
- How was containment verified?
Manual system: This requires managing spreadsheets that link every raw material to each production batch and finished device. You’ll need separate tabs for incoming materials, in-process assemblies, and shipments, all tied to batch or serial numbers.
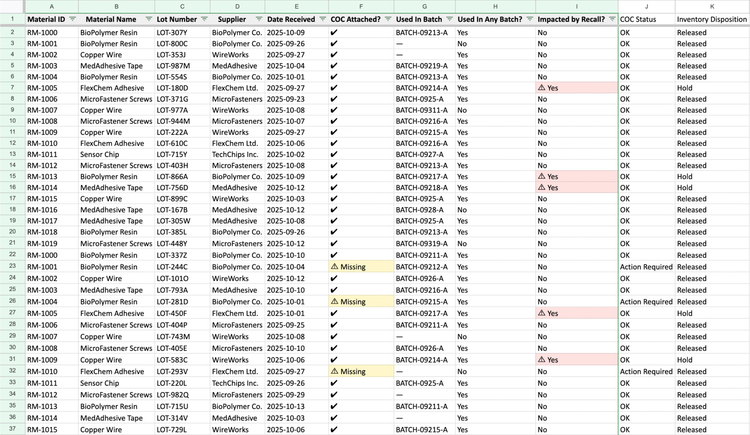
QMS software: Integrated systems like QT9 ERP and QT9 QMS take a strong approach to traceability. In the ERP, every part gets a lot number the moment it hits inventory, which links it to customer orders, materials used, and production dates.
The QMS pulls in version-controlled specs and SOPs that governed that specific batch so you have a complete paper trail.
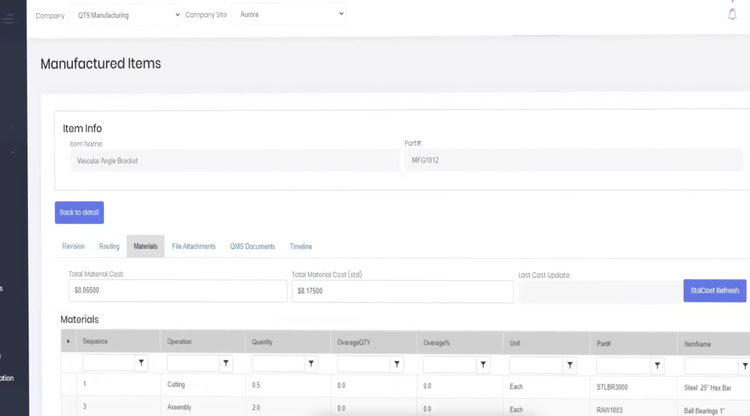
Supplier Management and Outsourced Processes
Traceability extends to supplier management, too. On paper, teams can show approved supplier lists and qualification records. Problems start popping up when a nonconforming part or recall forces auditors to ask how a supplier issue was identified, escalated, corrected, and verified.
Auditors need:
- Approved supplier lists
- Supplier qualification and requalification records
- Supplier-related nonconformances and corrective actions
Modern QMS platforms include supplier quality modules that link external issues directly to internal corrective actions.
Choosing the Best QMS Software
When we help medical device manufacturers choose QMS software for ISO 13485 compliance, the biggest concerns aren’t surface-level features. They’re wondering whether the system can actually hold up under audit pressure.
These are our top QMS software picks for medical device manufacturing:
Selecting ISO 13485 compliance software depends on:
- Team size and growth
- Design complexity
- Supplier risk exposure
- Traceability needs
- Audit frequency