FDA QMSR Explained: What Changed and What It Means for Your QMS
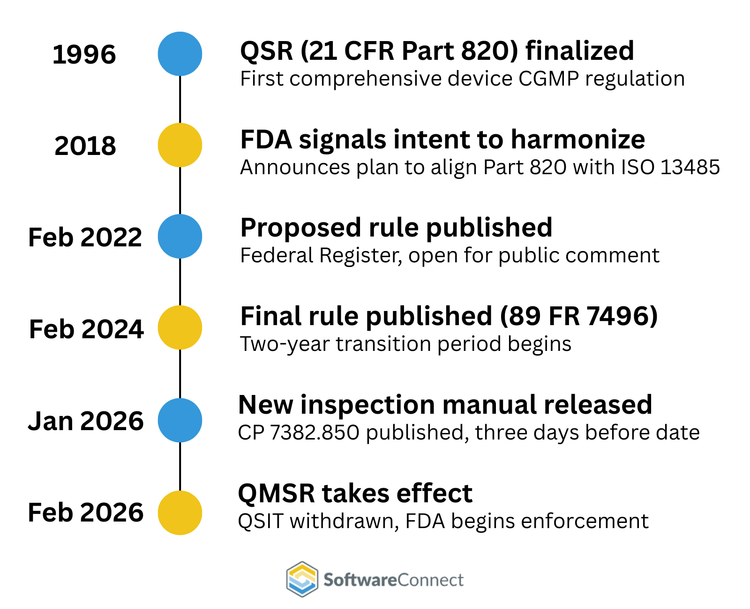
On February 2, 2026, the FDA’s Quality Management System Regulation (QMSR) officially replaced the Quality System Regulation (QSR) that had been in place for medical device manufacturing in the U.S. since 1996. The change is a significant milestone for medical device compliance. QMSR incorporates the international standard ISO 13485:2016 by reference into 21 CFR Part 820. This means all U.S. medical device manufacturers must now comply with an international quality management system standard, not just a domestic one.
If you manufacture finished medical devices for the U.S. market, QMSR applies to you. This page breaks down what actually changed, how it affects inspections and documentation, and what it means for your QMS software going forward.
Want to explore QMS options? Check out our best medical device QMS software page to compare top options.
QSR vs. QMSR: What Actually Changed?
QMSR replaces the bulk of the old Part 820 with a direct reference to ISO 13485. So now, instead of keeping its own requirements, the FDA defers to the ISO standard for most things. Most of the old QSR sections are now marked “Reserved,” meaning they’ve been withdrawn and held for potential further use. In their place, four new sections were added:
| Section | Title | What it Does |
|---|---|---|
| 820.7 | Incorporation by Reference | States that ISO 13485:2016 and ISO 9000:2015 Clause 3 are incorporated into Part 820. Provides instructions for obtaining the standards |
| 820.10 | Requirements for a QMS | The backbone of QMSR. Requires compliance with ISO 13485 AND additional FDA regulatory requirements (UDI per Part 830, MDR per Part 803, device tracking per Part 821). Extends implantable device traceability to life-sustaining devices. States that noncompliance renders a device “adulterated” and subject to regulatory action. |
| 820.35 | Control of Records | Defines specific record requirements for complaints, service records, UDI documentation, MDR/adverse events, and confidentiality provisions for records exchanged with the FDA. More detailed than the old QSR. |
| 820.45 | Device Labeling and Packaging Controls | Adds requirements beyond ISO 13485 Clause 7.5.1(e), including procedures for visual inspection of labels for UDI/UPC, expiration dates, storage, handling, and processing instructions. |
Risk-Based Thinking
One of the biggest changes isn’t a new section, but the way risk management is now throughout the entire QMS. Under the old QSR, risk was only explicitly addressed in design controls (Section 820.30). Under QMSR and ISO 13485, risk-based thinking runs through every process: supplier controls, production, CAPA, postmarket surveillance, and management review. This means manufacturers need documented risk rationale throughout their quality system, not just in design files.
Why the FDA Made This Change
The QSR hadn’t been significantly changed since 1996. And since then, ISO 13485 has undergone multiple revisions, eventually reaching the 2016 version. Because the two were very similar, it was redundant to have them both, so QMSR works to bridge the gap to make domestic and international regulatory expectations more similar. The main factors of the change are:
- Global Consistency: ISO 13485 is already used worldwide, and is the foundation for programs like the Medical Device Single Audit Program (MDSAP). Aligning Part 820 and ISO 13485 just reduces the burden for manufacturers selling in global markets.
- Cost Savings: The FDA estimates that QMSR will result in approximately $532 million in annual net cost savings for the medical device industry. This is based on eliminating redundant compliance efforts for manufacturers running systems to comply with multiple standards.
Key Terminology Changes
A lot of the changes to QMSR are semantic, and many of the familiar acronyms and terminology have changed to match ISO 13485. This matters because it affects SOPs, templates, training materials, and how you communicate during inspections. While using legacy terms is acceptable during the transition, you should start to transition to the new vocab.
| Old QSR Term | New QMSR/ISO 13485 Term | Notes |
|---|---|---|
| Device Master Record (DMR) | Medical Device File (MDF) | Requirements are substantially similar; ISO 13485 uses MDF as a catch-all for design and production documentation |
| Design History File (DHF) | Covered under MDF | No separate term; documentation requirements still apply under ISO 13485 Clause 7.3 |
| Device History Record (DHR) | Covered under MDF | Production records still required; terminology consolidated |
| "Safety and effectiveness” | “Safety and performance” | Aligns with ISO language. FDA clarifies this does not change the statutory standard under the FD&C Act. |
| “Establish” | Removed | ISO 13485 does not use this term; requirements are expressed differently |
| CAPA (combined) | Corrective Action/Preventive Action (separate) | ISO 13485 treats these as distinct processes in Clauses 8.5.2 and 8.5.3 |
Where ISO 13485 definitions conflict with FDA statutory definitions, the FDA’s definitions take precedence. For example, “device” and “labeling” as defined in the FD&C Act supersede their ISO 13485 counterparts.
FDA-Specific Requirements Beyond ISO 13485
It’s important to note that ISO 13485 certification does not equal QMSR compliance. This certification alone won’t exempt you from FDA inspections, mainly due to the FDA-specific additions that go beyond what ISO covers. These additional requirements include:
- Section 820.3 - Definitions: This retains and overrides certain FDA definitions that differ from ISO 13485 terminology. It also introduces five additional terms not found in either ISO 13485 or ISO 9000, like “component” and “finished device.”
- Section 820.35 - Control of Records: This requires specific documentation for complaint records, service records, UDI documentation per Part 830, MDR and adverse event records per Part 803, and confidentiality provisions governing records sent to and received from the FDA. This is a lot more specific than anything in the old QSR.
- Section 820.45 - Labeling and Packaging Controls: The FDA determined that ISO 13485 Clause 7.5.1(e) didn’t go far enough on labeling controls. This new section requires procedures for visual inspection of labels to verify the accuracy of UDI or UPC codes, expiration dates, storage and handling instructions, and processing instructions. This is in addition to existing requirements under 21 CFR Part 801.
- 21 CFR Part 4 – Combination Products: The final rule includes conforming edits to clarify QMS requirements for combination products. These edits align Part 4 with ISO 13485 and QMSR but do not change the existing CGMP requirements for combination products.
What Changed About FDA Inspections
While the regulation changes are significant, the inspection process has changed just as much. On February 2, 2026, the FDA retired the Quality System Inspection Technique (QSIT), which had been used since 1999, and replaced it with a new inspection process described in Compliance Program 7382.850.
The new manual replaces the old structure with a risk-based evaluation model that’s organized around six QMS areas and four Other Applicable FDA Requirements (OAFRs). The OAFRs are: Medical Device Reporting (803), Corrections and Removals (806), Medical Device Tracking (821), and Unique Device Identification (830). So instead of auditing predefined subsystems, investigators will focus on areas that pose the greatest risk to patients and device users.
So, what are the biggest changes? Several stand out:
- Items subject to FDA review: Management reviews, internal audit reports, and supplier audit reports are all now subject to FDA review. Under the old QSR, Section 820.180© explicitly exempted these records from FDA inspection, so you didn’t have to disclose these at all. This is big, because many companies use these documents to discuss sensitive subjects during internal audits, since they wouldn’t be seen. If that’s you, you’ll have to make sure they’re combed over before your next inspection.
- Remote Regulatory Assessments (RRAs) are now in play: The FDA can now assess your compliance through remote document review. This can be either ahead of or instead of on-site inspections. This gets interesting because if the FDA feels it can evaluate your quality system from documents and evidence alone, it may not send an inspector to your facility at all.
- Pre-QMSR records are not off limits: The FDA has clarified that investigators may review records before QMSR to help determine compliance with the new rule. The FDA recommends a comparative analysis or crosswalk showing how legacy records meet QMSR.
What Manufacturers Should Do Now
QMSR officially went into effect on February 2, 2026. So if you aren’t compliant now, you need to be ASAP. The FDA is enforcing these new requirements, so here are some of the main steps you need to follow.
- Conduct a QMSR-focused gap assessment. Map your current QMS against the new QMSR rule. Figure out where your documentation, processes, and risk management practices fall short and where you don’t comply. This is crucial for finding where you need to change.
- Update your documentation: Change your terminology from the old QSR language to the new one. Update your quality manual, SOPs, templates, and forms. Transition from DMR/DHF/DHR structures to the Medical Device File (MDF) framework.
- Train your teams: Every team member who is involved with quality processes needs to understand the new language, risk-based approach, and what inspectors will now be looking for. This can be ongoing as you refine your new workflows.
- Get on the same page as your suppliers: Supplier controls have a large emphasis in QMSR inspections, similar to MDSAP. Make sure your suppliers understand the new expectations, and get your audit and monitoring processes up to date.
How This Affects Your QMS Software
QMSR changes how your quality management system needs to operate going forward. If your current tools can’t adapt to the new structure, terminology, and documentation requirements, you should consider finding a new medical device QMS system. That’s because the broader scope of QMSR will expose limitations that may have been manageable under the old QSR. Here’s what to look out for when evaluating:
- ISO 13485-native workflows: Your QMS should be structured around ISO 13485 out of the box, not retrofitted from a generic platform.
- Medical Device File Support: The system should accommodate the consolidated MDF structure rather than forcing you into legacy DMR/DHF/DHR silos.
- Risk Management Traceability: Look for platforms that integrate risk management across design, production, and supplier management, not just design controls.
- Inspection-readiness features: Since management reviews, internal audits, and supplier audits are now subject to FDA review, your QMS needs to generate clean, retrievable reports for these activities on demand.
Here are some of the top medical device QMS software that will help you get QMSR compliant:
Greenlight Guru
Greenlight Guru is a QMS software that is built specifically for the medical device industry. It features full quality and clinical suites for not only medical device manufacturing, but for MedTech clinical studies as well. It’s a strong option for growing companies that need QMSR compliance.
QT9 QMS
QT9 QMS includes over 25 modules for several industries, including medical device manufacturers. It also has a full ERP that you can integrate to create an all-in-one system. It’s affordable, too, for small and growing businesses getting their first QMS software.
Qualio
Qualio is another system that’s made for the life sciences industry, including medical device manufacturers. It supports product development processes, compliance, and FDA audit readiness. The interface is also one of the most modern.
MasterControl
MasterControl Quality Excellence is built for medium to large-sized enterprises in highly regulated industries, including medical devices. The full QMS suite can handle larger data volumes and automates complex processes to help comply with QMSR.